Discover a portfolio of digital PCR products.
Digital PCR doesn’t have to be difficult to perform. Stilla Technologies has built a portfolio of products to help facilitate the detection and quantification of nucleic acids across applications spaces. From the Nio+ system, to easy-to-use mixes, to best-in-class consumables, to analysis software, Stilla makes digital PCR accessible to researchers and laboratories around the world.
Nio™+ System
Nio™+ is a versatile, all-in-one, fit-for-throughput, digital PCR instrument with unparalleled user-friendliness. The first dPCR system with 7-colors and continuous loading capabilities.
naica® system
The naica® system is the powerful, yet easy-to-use, multiplex digital PCR platform used for the precise and highly sensitive quantification of nucleic acids.
Digital PCR Kits and Assays
Get ready-to-use, fully-validated panels for the detection and quantification of nucleic acids.
Digital PCR Chips and Reagents
The naica® system’s digital PCR workflow is integrated into a single consumable chip, providing maximized sensitivity and throughput when combined with specially-formulated PCR reagents.
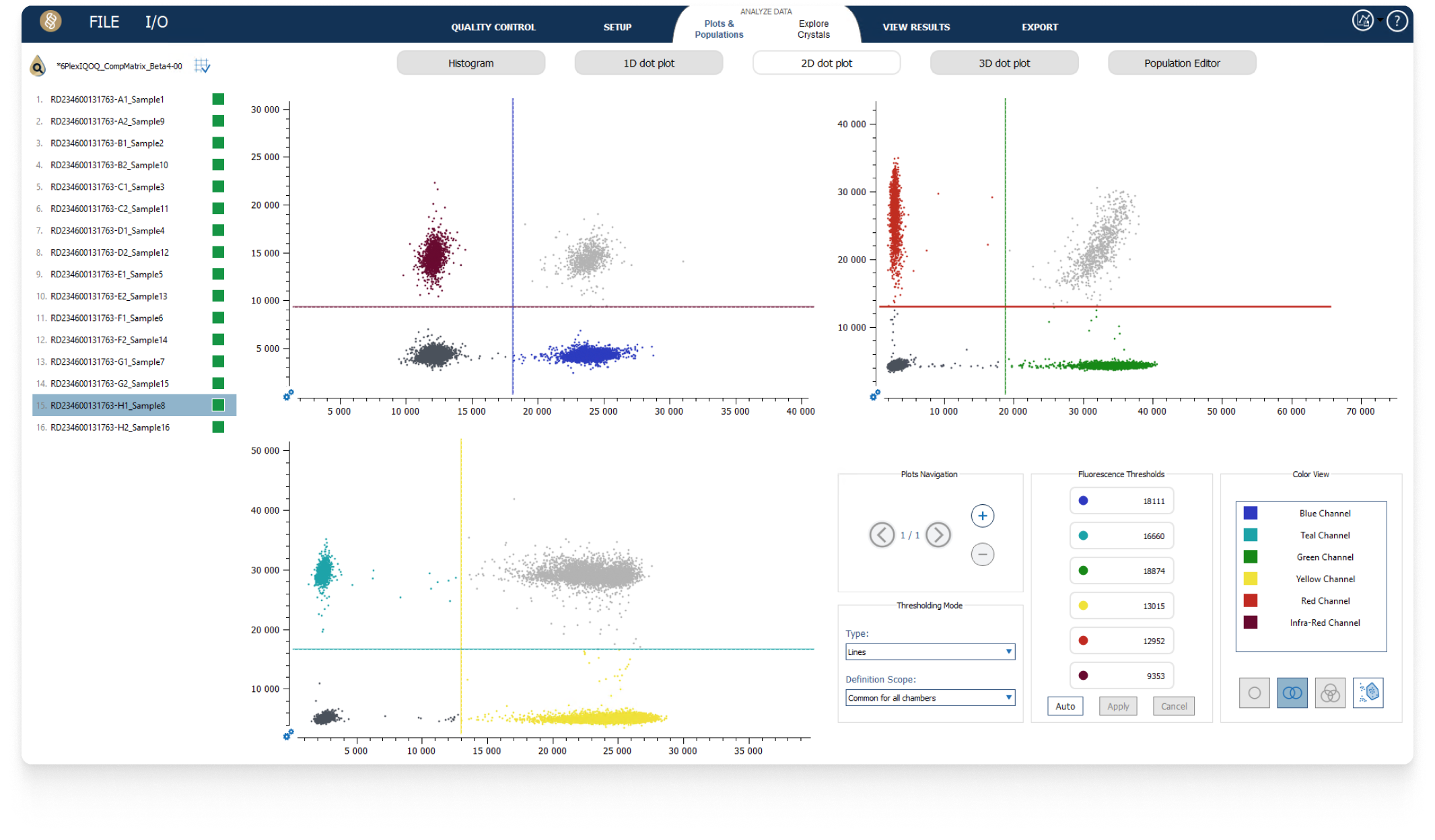
Digital PCR Analysis Software
Experience user-friendly software that provides intuitive visuals for image analysis.